文献翻译 --- 解析二倍体和多倍体基因组中的单倍型
解析二倍体和多倍体基因组中的单倍型
作者:Xingtan Zhang, Ruoxi Wu, Yibin Wang, Jiaxin Yu, Haibao Tang
原文来源:Zhang X, Wu R, Wang Y, Yu J, Tang H. Unzipping haplotypes in diploid and polyploid genomes. Comput Struct Biotechnol J. 2019; 18: 66 - 72.
关键字
基因组组装、单倍型定相、倍性、参考基因组、杂合性
摘要
二倍体基因组由两个同源的染色体拷贝组成,两个亲本各有一个,而多倍体基因组包含两个以上的同源染色体组。大多数参考基因组组装折叠了代表 “mosaic” 序列的单倍型,忽略了可能涉及重要细胞和生物学功能的等位基因变异。在最近的基因组研究中,将单倍型解析成不同的序列集已成为一种增长趋势,因为它是解决重要临床和生物问题(如复合杂合子、杂种优势和进化)的重要工具。这里,我们回顾了用于杂合二倍体和多倍体基因组的基于比对和组装的单倍型定相的现有方法,以及改进基因组定相的实验方法的最新进展。我们预计,在不久的将来,完整的单倍型定相可能成为基因研究中的常规程序。
1. 介绍
参考基因组的组装是当今许多生物提高遗传资源利用率的常用途径。随着单分子 long reads 测序的可用性,如:PacBio SMRT sequencing(单分子实时测序)、牛津纳米孔技术(ONT)、Bionano Genomics、以及高通量染色质构象捕获技术(Hi-C),与几年前相比,大多数基因组测序研究现在可以以较低的成本完成染色体水平的组装。
在大多数参考基因组组装中,通常每个染色体的两个同源拷贝,各来自不同的亲本,被折叠在一起,并被认为是当前二倍体基因组组装方法中两个单倍体的“mosaic”参考。这种参考以“单倍体”表示,它仅反映整个基因组中的单个单倍型。如,来自纽约 13 名志愿者的人类参考基因组(最新发布的 GRCh 38 )仅包含 ABO 血型基因座中的 O 等位基因。同样的,对于多倍体基因组,参考基因组中通常以单个单倍体为目标,从而忽略了测序生物体内的大量遗传多样性。
虽然“单倍体”参考更易于推理和比较,但它们通常无法获取生物体的二倍体或多倍体性质,从而忽略了可能具有潜在重要功能的等位基因变异。自二十年前人类基因组计划(HGP)初步完成以来,研究人员一直在尝试改进基因组组装,以在同一测序研究中完全解决这两种单倍型。最近测序技术的改进,特别是 long reads 测序技术的发展,为解决线性参考基因组中完全不存在的单倍型之间的结构变异提供了机会。在人类基因组研究中,两种单倍型的重建也具有临床相关性;如,当确定复合杂合突变的存在以及准确的人类白细胞抗原(HLA)分型时。
基因组定相的另一个重要应用是研究等位基因特异性表达(ASE)或等位基因失衡(AI),这已被认为是导致杂种优势的重要机制。ASE 或 AI 揭示了一种亲本等位基因优先于另一种表达的模式[1]。这种差异表达模式可能是由位于顺式调节元件中的等位基因变异引起的,它们能够与环境因素相互作用以调节复杂的表达网络[2],最终导致大的表型变异。例如,越来越多的证据表明,具有显性表达模式的转录更活跃的等位基因是杂交水稻杂种优势的重要贡献者[3], [4]。目前识别 ASE 的方法依赖于将 RNA-Seq reads 定相到不同的单倍型上。通常,在 Illumina short reads 测序平台上产生的 RNA-Seq reads 与参考基因组比对,属于每个亲本基因组的变异被定相,相应的 reads 进一步用于量化其各自等位基因的基因表达[3]。
此外,等位基因变异的鉴定还可以促进多倍体进化的研究,并为作物育种提供信息。复杂的多倍体甘薯基因组首先被组装成一个共有参考基因组,一种新的单倍型定相方法成功地产生了一个单倍型水平的基因组组装[5]。使用分型等位基因变异的系统发育分析共发现了六种单倍型,从而追踪了甘薯谱系中的六倍体化历史[5]。在甘蔗项目中,我们最近开发了一种新的 ALLHiC 算法,该算法能够通过结合 PacBio long reads 测序和 Hi-C 技术来构建多倍体甘蔗基因组的等位基因感知和染色体水平组装[6],[7]。具有单独组装的单倍型的分型甘蔗基因组进一步揭示了甘蔗谱系中复杂的染色体重排和进化历史[6]。
单倍型定相已成为杂合和多倍体基因组组装中的一个基本问题。单倍型定相是准确地表示给定生物体遗传组成的关键。在这里,我们回顾了现有的单倍型定相计算方法 —- 包括杂合和多倍体基因组的基于比对和基于组装的定相方法。两种定相方法的概述如 图1 所示。我们通过对实验进展的简要总结来跟进计算方法的讨论,以帮助基因组定相。最后,讨论了当前方法的局限性以及未来的方向。
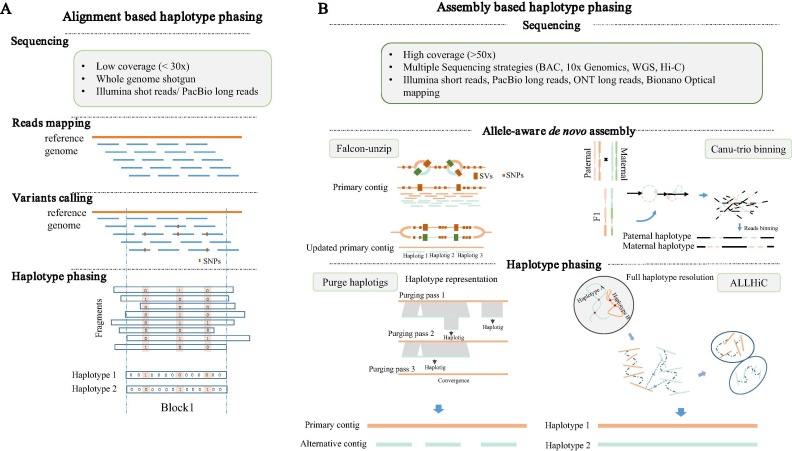
图 1 . 两大类单倍型定相策略概述。左图 (A) 基于比对,右图(B)是基于组装的单倍型定相流程。在基于比对的单倍型定相中,reads 以相对较低的覆盖率(< 30X)进行测序,并比对到参考基因组以识别变异位点。接着,连锁变异被扩展为不同的 phased blocks ,每个 block 含有许多相邻的 SNPs,表示为 0(REF)/1(ALT) 。(B)图显示基于组装的单倍型定相中,通常使用各种测序技术进行更深层次的测序。可以使用 Falcon-unzip 或 Canu-trio binning 方法实现等位基因感知的从头组装。当组装多个单倍型时,可以选择主要 contigs 作为任意单倍型表示,例如,使用 purge_haplotigs 进行下游分析。或者,可以通过 Hi-C 技术解析全部单倍型,如使用 ALLHiC。
2. 基于比对的单倍型定相
下一代测序(NGS)成本的大幅下降为全基因组规模的单倍型定相提供了大量数据,并促进了过去二十年中定相算法的不断发展。当参考基因组可用时,最直接的单倍型定相方法是先与全基因组测序(WGS)reads 进行比对,然后在多态位点编译一组杂合基因组,最后连续配对相邻的单倍型(图 1)。相邻位点的准确配对信息由同一 WGS reads 或 reads 双端上的等位基因共现提供。
基于不同的优化标准或目标函数设计了不同的基于对齐的定相算法,包括最小误差校正(MEC)、甲醛最小字母翻转(WMLF)和最大片段切割(MFC)[8]。这些优化标准通常是 NP -hard,因此通常依赖于启发式方法来加速计算。简而言之,MEC 在于通过应用最少数量的碱基校正来重建两种单倍型[9]。WMLF 是从 MEC 算法改进得来,通过基因翻转次数来测量误差[8]。MFC 将单倍型确定问题转换为 Max-Cut 问题,并通过搜索可以链接单倍型的每个 SNP 边缘的最大距离来找到解决方案。其他算法,如 图10,启发式动态规划[11],混合模型[12]和模糊冲突图[13],也适用于基因组定相,每种算法都利用不同的统计模型来处理错误。
2015 年 Je-Keun 等人[8] 已经审查了许多基于这些算法的单倍型定相方法。因此,我们只关注自 2015 年以来新开发的基于 NGS 的分型算法,并强调它们各自的优势和局限性(表1)。WhatsHap 提供了一种动态规划算法来解决 wMEC 问题,这使得运行时复杂度于 SNP 的数量成线性关系[14]。HapCUT2 是 HapCUT 程序的扩展,能够处理广泛的测序技术,包括 NGS short reads、long reads、 linked-reads 和 Hi-C reads。与 HapCUT 不同,HapCUT2 采用基于似然的模型来估计技术特定的错误,并在读取单倍型图中使用最大切割计算迭代地搜索变体子集[15,p.2]。SHAPEIT 是一系列软件程序,在于根据群体水平的多态性数估计单倍型。最近开发的 SHAPIT3 修改了马尔科夫链蒙特卡洛(MCMC)采样程序,加快了该过程,并且能够以非常低的单倍型转换错误率处理生物样本库规模的数据集[16]。作为 SHAPEIT3 的扩展,SHAPEIT4 结合了基于位置 Burrow Wheeler 变换(PBWT)的方法,可从参考 panel 中快速选择一小组信息丰富的单倍型[17]。SHAPEIT4 还表现出随样本大小的亚线性缩放,并允许整合外部定相信息,如单倍型的大型参考 panel、预定相变体的集合和长测序 reads[17]。
表 1. 本研究中列出的软件概述。
| 程序 | 算法 | 输入数据 | 强调 | 局限性 | 引文 |
|---|---|---|---|---|---|
| 基于比对的单倍型定相 | |||||
| WhatsHap | 动态规划算法 | VCF/BAM/reference genome | 在完整性和准确性方面表现良好,适用于短读和长读 | 忽略结构变化,只允许输入数据的最大覆盖率为 15x;仅适用于二倍体基因组 | [14] |
| HapCUT2 | 基于 MAX-CUT 的启发式算法 | VCF/BAM/reference genome | 能够处理各种测序技术,包括 Illumina short、PacBio long、10x Linked 和 Hi-C reads | 忽略结构变化,仅适用于二倍体基因组 | [15] |
| SHAPEIT3&4 | HMM/MCMC/PBWT | VCF/Genetic map | 在准确性和速度方面表现出色,能够处理大数据量 | 忽略结构变化,不适合直接定相低覆盖深度的测序数据 | [16], [17] |
| 基于组装的单倍型定相 - 单倍型表示 | |||||
| HaploMerger1/2 | Whole genome comparison | Draft genome assembly | 适用于杂合度高的二倍体基因组组装;实施灵活灵敏的组装错误检测 | 不适合过于碎片化的 scaffold (e.g. N50 < 100 kb) | [26], [27] |
| Redundans | Whole genome comparison | Draft genome assembly | 多种功能,包括去除杂合序列、scaffolds、间隙闭合 | 可能会在 reducing 步骤中丢弃一些重复和旁系同源的 contigs | [23] |
| Purge_haplotigs | Read depth | BAM/draft genome assembly | 它能够避免部分重复和旁系 contigs 被过度清除;非常快并且可以很好地扩展到大的基因组大小 | 由于 contigs 的任意保留,无法解决基因组草图中的单倍型转换,“伪单倍体”并不代表多态性区域中的真正相位 | [28] |
| FALCON&FALCON-Unzip | 识别 ‘bubble’ 结构的启发式算法和辅助构建单倍型的贪心算法 | PacBio raw reads | FALCON-Unzip 能够组装高度准确、连续的主要 contigs 和单倍体,从而允许在单倍型水平进行进一步的下游分析 | Primary contigs 包含相邻 “phase blocks”之间的单倍型转换错误;重复序列和杂合序列的检测会相互干扰,导致错误的单倍型组装 | [19], [20] |
| CANU | Trio binning | Parental short reads/F1 long reads | 能够为每一个亲本生成两组单倍体基因组;Trio binning 在连续性和准确性方面表现出色 | 没有记录谱系信息的高度杂合基因组的有限应用 | [21] |
| 基于组装的单倍型定相 - 完整的单倍型分辨率 | |||||
| FALCON-Phase | 一条整合 PacBio reads 和 Hi-C 数据的管道,为二倍体基因组重新分配单倍型 | Output for FALCON-unzip assembly/Hi-C reads | 可以从 FALCON-Unzip 的等位基因感知 contigs 组装中受益;整合 PacBio contigs 组装和 Hi-C reads | 仅适用于二倍体基因组;与其他 contigs 组装工具不兼容 | [36] |
| ALLHiC | Prune/optimize/Genetic Algorithm | Draft contig assembly/Hi-C mapping BAM/Allele table | 适用于各种复杂程度不同的基因组,包括简单二倍体、杂合二倍体、异源多倍体基因组和自身多倍体基因组;当 contigs 连续性较低时性能更好 | 对起始 contigs 组装的准确性敏感;需要密切相关的参考基因组来生成等位基因重叠群表 | [7] |
3. 具有单倍体代表的基于组装的单倍型定相
与主要针对小变异的基于比对的定相方法相比,基于组装的方法通常更加准确,并且可以覆盖更大类型的基因组变异,如,大插入缺失和结构变异(表1)。然而,由于存在多个单倍型,杂合二倍体或多倍体基因组组装可能难以组装,导致初始 contig 水平组装中的歧义和冗余。
为了处理这些歧义和冗余,组装杂合基因组的常见做法是简单地重建单个单倍型来代表整个基因组(图1)。杂合基因组的组装可能导致在 contig 水平组装中分离的具有高水平差异的序列,即在同源染色体的相同基因座处存在不同的等位基因。由于单倍型之间的这些共同序列,初步 contig 水平组装通常包含亲本等位基因之间超过 90% 相似的序列。输入 contigs 的成对基因组比对可用于确定冗余 contigs,通过比较初步的 contigs ,其代表来自多态性区域的不同单倍型。该方法已在 Redundans 计划 [23] 中实施,并成功辅助杂合基因组组装,如,Echinochloa crus-galli [24] 和 Colorado potato beetle[25]。
另一个自动化流程 “HaploMerger” 被提出来重建二倍体组装中 contigs 的等位基因关系[26]。HaploMerger 采用 LASTZ-ChainNet 方法进行全基因组比较,采用所谓的二倍体基因组组装(DGA)图来描述二倍体基因组组装中中间基因或同源物之间的关系[26]。HaploMerger 在其测试过程中在几个多态性二倍体基因组上表现出优异的性能,并且没有引入新的组装错误,显示出其在分析和利用多态性基因组组装方面的功效。HaploMerger2(HM2)是旧流程的重大升级,重新设计了从短读和长读二倍体组装的单倍型重建[27,p.2]。HM2 可以处理低杂合和高杂合组装,还提供更灵活的组装错误检测和可靠的间隙闭合方法,从而比旧流程大大提高最终二倍体水平组装的连续性[27, p.2]。
除了全基因组比对方法外,基于 reads 测序深度的方法还可用于确定杂合二倍体基因组组装中的等位基因重叠群。对于具有高水平杂合性的二倍体基因组组装,预期 reads 深度的双峰分布构成对不同来源的 contigs 进行分类的基础。“Purge Haplotigs” 程序能够利用单个 contigs 的 reads 深度来识别疑似重复序列的 contigs ,这些重复序列被假定为等位基因单倍型,需要重新分配以获得简单的“伪-单倍体”[28]。虽然推导相对简单,但重要的是要注意这种“伪-单倍体”并不代表杂合基因组的真正定相,因为选择保留哪些 contigs 仍然是任意的。
4. 具有完整单倍型分辨率的基于组装的单倍型定相
虽然单个单倍体表示在杂合组装中相对简单,但它也丢失了大量属于其他单倍体的序列信息。现代测序方法,如 Pacific Biosciences 和 Oxford Nanopore Technology (ONT) 开发的单分子实时测序(SMRT)可提供long reads,有望在复杂基因组中实现压倒性的组装性能[18](图1)。特别是,long reads 比short reads 有望恢复更长的单倍型。
目前,Pacific Biosciences 技术经常用于组装大量植物基因组,但迄今为止完成的大多数基因组组装都集中在纯合个体或近交系,产生单一的代表性单倍型。然而,对于许多难以作为纯合个体进行近交的植物作物,例如许多热带水果作物,杂合个体的组装需要格外小心。为了解决这个问题,开发了几种算法在从头基因组组装过程中重建多个单倍型。这种算法试图超越基因组过于简单的单倍型表示,这对于杂合二倍体和多倍体生物特别有价值。
FALCON 和 FALCON-Unzip 算法提供了一种干净的解决方案,可以从头组装原始测序数据并持续识别定相二倍体基因组[19]。首先,FALCON 从校正后的 PacBio 序列数据中读取序列比对,然后基于 read overlaps [20] 构建字符串图。在此过程中,字符串图通常包含多组 “haplotype fusion”,它们是重叠的读取组,在图中显示为 “bubbles”。“bubbles” 代表同源序列之间的主要结构变异和高度不同的区域。为了解决组装图中的这些 “bubbles”,FALCON-Unzip 分析了单倍型融合组,并找到了混合变体作为 “unzip” 其他融合单倍型的基础。一个单倍型,通常是随意的,首先被确定为图中的主要路径,或 “primary” contigs;而另一种代表替代路径的单倍型被称为 “associated” contigs。该方法用于从 Columbia-0 (col-0) 和 Cape Verde Island-0 (Cvi-0) 拟南芥生态型,Vitis vinifera CV(赤霞珠)和高度杂合的野生二倍体 Clavicorona pyxidate [19] 重新组装 F1 基因组。这些单倍型解析的组装反映了它们各自基因组更真实的表示,并允许以更好的准确性和分辨率研究单倍型结构和杂合性。
一种更新的方法,“trio binning”,通过在组装前解决等位基因变异来简化单倍型组装[21],该方法已在 CANU 组装工具[22]中实现。与现有方法相比,该方法的有效性随着杂合度的增加而增加。Trio binning 首先将来自后代的长 reads 划分为单倍型特定组,由两个父母中的每一个基因组测序指导。分割后,每个单倍体独立组装,从而完成完整的二倍体重建。为了说明“trio binning”的实用性,对牛亚种 Bos taurus taurus 和 Bos taurus indicus 之间的 F1 杂交种进行测序并完全分解为两个亲本单倍型,单倍体 NG50 大小超过 20 Mb,准确率超过 99.998%,超过当前牛参考基因组的质量[21]。虽然准确度高,但 “trio binning” 的设置需要预先知道亲本并对其进行测序,以便在 F1 基因组中进行分组。这一要求不仅会导致更高的成本,还在某些情况下受到限制,如当植物物种是从野外采集或育种计划欠发达时,可能无法获得亲本信息。
5. 利用 Hi-C 技术重建染色体水平的单倍型
结合 PacBio long reads 测序和基于邻近连接的基因组组装是构建染色体水平基因组组装的有效方法。高通量染色质构象捕获(Hi-C)是一种源自染色质构象捕获(3C)的技术,结合了染色质邻近连接方法和高通量测序,从而获得整个染色体上染色质相互作用的精细图谱[29]。交联染色质通过限制性内切酶切割,接近原位连接以获得相互作用的 DNA 片段。连接的 DNA 片段被生物素捕获,然后通过双端测序进行测序。相互作用的 DNA 片段显示为成对的连锁 reads,揭示了有关整个染色体中序列的分组和线性组织的长距离信息[30]。染色体内接触的概率随着线性距离呈现指数级衰减,但与同一染色体(染色体间)上的位点相互作用的概率仍然比不同染色体(染色体间)上的位点的概率高得多,即使在同一染色体上相隔超过 200Mb ,也可能产生相互作用[31]。基于邻近连接信息,Hi-C 数据可以有效的识别 contigs 或 scaffolds 之间的连锁,使 contigs 几乎与整个染色体尺度相关联。
在过去的十年中,基于 Hi-C 的组装方法已经广泛应用于哺乳动物、植物和昆虫中,从而生成可靠的染色体水平的从头组装基因组。Hi-C 数据也可用于将基因组分型到染色体水平的单独单倍型上,因为同源染色体在细胞核中占据不同的区域[32],这可用于区分不同的单倍型。因此,基于这些不同的 DNA 结构域,使用基于 Hi-C 方法的单倍型感知染色体水平组装已经发表并成功应用于几个复杂的多倍体基因组,如普通小麦[33],花生[34]和棉花[35]基因组。
最近发布的一个基因组 phasing 和 scaffolding 软件是由 Phase Genomics 开发的 FALCON-Phase[36]。FALCON-Phase 能够通过整合二倍体个体的 long reads 测序数据和 Hi-C 数据,将 contigs 或 scaffolds 定相连接到高质量的单倍型基因组组装上[36]。该管道建立在前面提到的 FALCON-Unzip 的基础上,可生成 phased blocks ,并减少单倍型交换错误。FALCON-Phase 可以解决 switches 和 backfill 纯合子区域的问题,以产生染色体水平的分型二倍体基因组组装。
然而,FALCON-Phase 管道是为了定相二倍体基因组而设计的,尚不支持多倍体基因组染色体的构建。为了解决这一限制,专门为多倍体基因组开发了一种名为 ALLHIC 的新 Hi-C scaffolds 管道[7]。ALLHIC 使用新的 pruning 步骤来去除等位或交叉等位基因的 Hi-C 连接(不同单倍型之间的连接)。这样的连接在定相时通常是有问题的,因为它们通常会阻碍单倍型单独分型。因此,ALLHiC 能够将 Hi-C 数据中的超长单倍体信息与高质量的初步组装相结合,从而实现同种多倍体和杂合二倍体基因组的准确分型,以构建染色体规模的单倍型组装体。它已被成功应用于几个染色体规模的同源多倍体基因组,包括同源四倍体[6]和同源八倍体甘蔗基因组[7]。除了同源多倍体基因组外,ALLHiC 适用于广泛的基因组,包括简单二倍体、杂合二倍体和异源多倍体基因组。
6. 针对单倍型分辨基因组测序的实验方法
最初的人类基因组计划主要是通过对插入细菌人工染色体(BAC)中的长 DNA 片段(50-200kb)的大插入克隆进行分级测序[37]。类似的,fosmids 也可以用作 DNA 载体,因为它利用 F 质粒复制起点和分配机制来克隆大的 DNA 片段。fosmid 文库的测序已成功应用于高度杂合的基因组,如杂合的小菜蛾基因组[38]。Fosmid 和 BAC 克隆代表了一种 ”divide-and-conquer“ 的方法,通过将基因组分解成更容易组装但仍保留单倍型信息的 block,因为每个单独的克隆代表一个单倍型。
Linked-Reads 是 10x Genomics 开发的一种全新的测序技术,有望以相对较低的成本进行全基因组单倍型定相。该技术利用微流体对高分子 DNA 分子进行分区和 barcode 化,然后对其进行 illumina short reads测序。包含相同 barcode 的双端 reads 被认为来自相同的单倍型,并在基因组组装过程中连接在一起,这大大降低了基因组组装的复杂性。使用 Linked-Reads 技术,最近一项关于人类基因组的研究揭示了不同人群中替代单倍型的综合列表[39]。
另一种相关技术,北京基因组研究所(BGI)开发的单管长片段 reads (stLFR)技术,最近显示了其在单倍型分型和从头组装方面的潜力[40]。stLFR 文库的构建涉及将转座子整合到 DNA 分子的长片段中,这些片段进一步与含有共享接头的珠子混合。在基因组 DNA 被捕获到磁珠上后,转座子与 barcode 接头连接,然后对 co-barcoded 的 DNA 片段进行双末端 reads 测序[40]。
克隆、连锁 reads 和长片段 reads 都提供了数百 kb 碱基到几兆碱基长度范围内的远程连锁证据,以支持单倍型的定相 —- 远远超出单个 WGS reads 提供的范围,包括最新的 nanopore reads。基于比对和基于组装的方法都能够在基因组定相期间利用这些实验证据。定相实验方法的流行很大程度上取决于它们各自的通量和成本。
7. 总结与展望
尽管现在有许多方法,包括计算方法和实验方法,但实现完整和完全准确的单倍型定相仍然具有挑战性。目前,我们回顾了两种单倍型定相的总体策略(图 1)。第一类单倍型定相方法依赖于全基因组测序 reads 的变异识别。如果单个二倍体或多倍体基因组中的杂合变体存在于相同的 reads 或者连锁的 reads ,则它们被分类到相同的 block 中。我们将这类定相策略称为基于比对的单倍型定相,它仅显示通常是小变异的定相等位基因变体,如 SNPs 或小 indels。由于基因组比对中经常遗漏来自较大变异的 reads ,因此基于比对的定相方法无法恢复较大的插入缺失或结构变异,如已知结构多样的人类 HLA 基因座或植物 S 基因座。第二类单倍型定相是基于组装的单倍型构建。对于这种策略,产生了单个单倍型的全长序列,由于提供的分辨率高于基于比对的定相,因此通常是首选的定相方法。
目前,大多数最先进的算法采用基于比对的策略来确定基于 illumina short reads 和/或 PacBio long reads [8]的相邻基因座,如 HAPCUT2[9]、WHATSHAP[14]和 WinHAP2[41]。然而,使用 illumina short reads 进行定相通常会产生有限长度的 phased blocks,因为相邻多态位点的距离可能超过典型 illumina reads 或 reads pair 的长度。尽管 long reads 测序技术在某些情况下能够扩展单倍型块,但它存在大量的测序错误,导致在同一 block 中混合等位基因信息的高水平 switch errors 。
对于基于组装的策略,新一代基因组组装工具(如 FALCON-Phase 和 ALLHiC)能够分离等位基因 contigs, 并为杂合二倍体或多倍体基因组生成染色体水平的基因组组装[12],[7]。然而,在目前的实验中,我们仍然有高水平的 chimeric contig assembly或 collapsed sequences,这可能导致大部分错误连接的 scaffolds,甚至导致染色体缺失[7]。目前,这两种策略都不是完美的,并且在单倍型重建过程中都可能导致很大一部分错误。
需要进一步发展单倍型定相和单倍型分辨基因组组装技术[42]。最近,Pacific Biosciences 推出了新的 Sequel II 系统,其通量相较于之前的 Sequel 系统高得多。这个新系统能够提供高度准确的单个长 reads (HiFi reads),利用 long reads 技术大大提供了准确性(超过 99.9% 的准确性)。使用 HiFi reads 对人类基因组进行从头组装产生了连续且准确的基因组,其 contig N50 超过 15Mb,一致性准确度为 99.997%[43]。随着类似的新测序技术的出现以及基因组组装算法的相应进步,杂合基因组和复杂多倍体基因组的完整单倍型解析可能很快成为基因组研究中的常规程序。
竞争利益声明
作者声明,他们没有已知的可能会影响本文报道的工作竞争性经济利益或个人关系。
致谢
这项工作得到了国家重点研发计划 (No. 2016YFD0100305 to H.T.)、国家自然科学基金 (No. 31701874 to X.Z.)、福建省自然科学基金 (No. 2018J01604 to X.Z.) 、闽台作物生态害虫防治国家重点实验室(No.SKL2018001 to X.Z.) 的支持。
附录 A. 补充数据
本文的补充数据可以在 https://doi.org/10.1016/j.csbj.2019.11.011 在线找到。
参考文献
[1] Gaur U, Li K, Mei S, Liu G. Research progress in allele-specific expression and its regulatory mechanisms. J Appl Genet 2013;54(3):271–83.
[2] Knowles DA et al. Allele-specific expression reveals interactions between genetic variation and environment. Nat Methods 2017;14(7):699–702.
[3] Huang X et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat Commun 2015;6(1):6258.
[4] Shao L et al. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis. Proc Natl Acad Sci 2019;116(12):5653–8.
[5] Yang J et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat Plants 2017;3(9):696–703.
[6] Zhang J et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat Genet 2018;50(11):1565–73.
[7] Zhang X, Zhang S, Zhao Q, Ming R, Tang H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 2019;5(8):833–45.
[8] Rhee J-K, Li H, Joung J-G, Hwang K-B, Zhang B-T, Shin S-Y. Survey of computational haplotype determination methods for single individual. Genes Genomics 2016;38(1):1–12.
[9] Bonizzoni P, Dondi R, Klau GW, Pirola Y, Pisanti N, Zaccaria S. On the minimum error correction problem for haplotype assembly in diploid and polyploid genomes. J Comput Biol 2016;23(9):718–36.
[10] Duitama J, Huebsch T, McEwen G, Suk E-K, Hoehe MR. ReFHap: a reliable and fast algorithm for single individual haplotyping. In Proceedings of the first ACM international conference on bioinformatics and computational biology - BCB ’10, Niagara Falls, New York, 2010, p. 160.
[11] Xie M, Wang J, Jiang T. A fast and accurate algorithm for single individual haplotyping. BMC Syst Biol 2012;6(Suppl 2):S8.
[12] Matsumoto H, Kiryu H. MixSIH: a mixture model for single individual haplotyping. BMC Genomics 2013;14(S2):S5.
[13] Mazrouee S, Wang W. FastHap: fast and accurate single individual haplotype reconstruction using fuzzy conflict graphs. Bioinformatics 2014;30(17): i371–8.
[14] Patterson M et al. WHATSHAP: weighted haplotype assembly for futuregeneration sequencing reads. J Comput Biol 2015;22(6):498–509.
[15] Edge P, Bafna V, Bansal V. HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 2017;27(5):801–12.
[16] O’Connell J et al. Haplotype estimation for biobank-scale data sets. Nat Genet 2016;48(7):817–20.
[17] Delaneau O, Zagury J-F, Robinson MR, Marchini J, Dermitzakis E. Integrative haplotype estimation with sub-linear complexity. Bioinformatics, preprint, Dec. 2018.
[18] Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinf 2015;13(5):278–89.
[19] Chin C-S et al. Phased diploid genome assembly with single-molecule realtime sequencing. Nat Methods 2016;13(12):1050–4.
[20] Myers EW. The fragment assembly string graph. Bioinformatics 2005;21 (Suppl). pp. 2, pp. ii79–ii85.
[21] Koren S et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat Biotechnol 2018;36(12):1174–82.
[22] Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Res 2017;27(5):722–36.
[23] Pryszcz LP, Gabaldón T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res 2016;44(12). e113–e113.
[24] Guo L et al. Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nat Commun 2017;8(1):1031.
[25] Schoville SD et al. A model species for agricultural pest genomics: the genome of the Colorado potato beetle, Leptinotarsa decemlineata (Coleoptera: Chrysomelidae). Sci Rep 2018;8(1):1931.
[26] Huang S et al. HaploMerger: reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res 2012;22(8):1581–8.
[27] Huang S, Kang M, Xu A. HaploMerger2: rebuilding both haploid subassemblies from high-heterozygosity diploid genome assembly. Bioinformatics 2017;33(16):2577–9.
[28] Roach MJ, Schmidt SA, Borneman AR. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinf 2018;19 (1):460.
[29] Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009;326 (5950):289–93.
[30] van Berkum NL, et al. Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp May 2010;39:1869.
[31] Schmitt AD, Hu M, Ren B. Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol 2016;17(12):743–55.
[32] Korbel JO, Lee C. Genome assembly and haplotyping with Hi-C. Nat Biotechnol 2013;31(12):1099–101.
[33] Maccaferri M et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat Genet 2019;51(5):885–95.
[34] ZhuangWet al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat Genet 2019;51 (5):865–76.
[35] Hu Y et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat Genet 2019;51(4):739–48.
[36] Kronenberg ZN, et al. Extended haplotype phasing of de novo genome assemblies with FALCON-Phase. Genomics, preprint, May 2018.
[37] International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature Feb. 2001;409(6822):860–921.
[38] You M et al. A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet 2013;45(2):220–5.
[39] Wong KHY, Levy-Sakin M, Kwok P-Y. De novo human genome assemblies reveal spectrum of alternative haplotypes in diverse populations. Nat Commun 2018;9(1):3040.
[40] Wang O et al. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res 2019;29(5):798–808.
[41] Pan W, Zhao Y, Xu Y, Zhou F. WinHAP2: an extremely fast haplotype phasing program for long genotype sequences. BMC Bioinf 2014;15(1):164.
[42] Schatz MC, Cosgrove A. Graph genomes article collection. Genome Biol 2019;20(1).
[43] Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 2019.