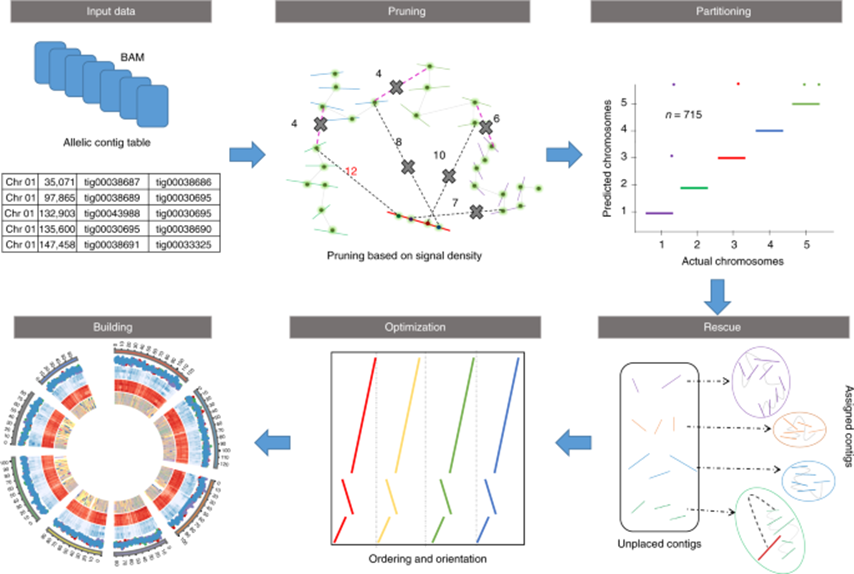
2019年8月5日,福建农林大学基因组中心张兴坦副教授和唐海宝教授研究组在Nature Plants杂志在线发表题为“Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data”的研究论文,开发了一种 ALLHiC 算法,成功完成了同源四倍体和同源八倍体甘蔗染色体水平组装。ALLHiC 可以从头组装获得单倍型染色体水平的多倍体基因组,分离每一个等位基因。ALLHiC 同时适用于多倍体和杂合二倍体的单倍型染色体水平组装。
作者对 ALLHiC 所有代码进行开源
Github:tangerzhang/ALLHiC: ALLHiC: phasing and scaffolding polyploid genomes based on Hi-C data (github.com)
Pipeline:Home · tangerzhang/ALLHiC Wiki (github.com)
Parameters: -h : help and usage -i : Allele.ctg.table ## 等位基因重叠群表,内含等位基因对信息 -b : input bam file -r : draft.asm.fasta
Tips: Details on how to identify allelic contigs can be found in the following link: https://github.com/tangerzhang/ALLHiC/wiki/ALLHiC:-identify-allelic-contigs
Parameters: -h : help and usage. -b : prunned bam (optional, default prunning.bam) -r : draft.sam.fasta -e : enzyme_sites (HindIII: AAGCTT; MboI: GATC) ## 限制性酶切位点 -k : number of groups (user defined K value) ## 可以设置多个 K 值,从而比较出最佳结果 -m : minimum number of restriction sites (default, 25)
Parameters: -h : help and usage. -b : sample.clean.bam (unpruned bam) -r : draft.sam.fasta -c : clusters.txt -i : counts_RE.txt -m : minimum single density for rescuing contigs (optional, default 0.01) ## 这里设置保留 contigs 的最小阈值,默认是 0.01 Tips: clusters.txt (-c) and counts_RE.txt (-i) can be generated by allhic extract. The format of clusters.txt is like below: #Group nContigs Contigs 48g1 506 tig0000150 tig0000151 tig0000152 48g2 692 tig0000097 tig0000114 tig0000231 48g3 683 tig0000015 tig0000035 tig0000235 ## 组别 该组含有的congtis个数 具体的 contigs 编号 The first column is group name. The second is number of contigs anchored in the group and the third lists contigs in the group.
allhic extract sample.clean.bam draft.asm.fasta --RE AAGCTT allhic optimize group1.txt sample.clean.clm allhic optimize group2.txt sample.clean.clm ... allhic optimize group16.txt sample.clean.clm ## optimize 需要运行 -k 16 次;故这里我们可以添加一个循环,以便自动运行 16 次 groups=16 for i in {1..$gourps}; do allhic optimize group${i}.txt sample.clean.clm; done
Parameters:
allhic extract Input files: bam file and contig-level assembly ## bam 文件和 contig 水平的组装 fasta 文件 Options: --RE value Restriction site pattern (default: "GATC") ## 限制性酶切位点
allhic optimize Input files: **counts_RE.txt - counts of restriction sites for each ## 在每组中对酶切位点的数量统计 contig in a clusterd group, which can be generated ## 以及 contig 长度 by allhic extract. The format is like below: #Contig RECounts Length tig0000001 572 157863 tig0000002 143 33000 tig0000003 231 60910 tig0000004 3789 1044000 tig0000005 646 166098 tig0000006 67 15000 tig0000007 1094 319000 **clmfile - The file records basic information of Hi-C ## 该文件记录了两个 contigs 之间 Hi-C 链接的 links between two contigs, including potential ordering ## 基本信息,包含顺序和方向 and orientation, number of supported reads and distance of ## 由 allhic extract 生成 paired-end reads. This file can be accessed by allhic extract. Options: --skipGA Skip GA step ## 跳过 GA 过程 --resume Resume from existing tour file ## 从现有的 tour 文件中恢复 --seed value Random seed (default: 42) ## 随机种子? --npop value Population size (default: 100) --ngen value Number of generations for convergence (default: 5000) ## 收敛代数 --mutpb value Mutation prob in GA (default: 0.2) ## 遗传算法(GA)中的突变概率
Build
将 tour 格式的文件转换成 fasta 序列文件和 agp 位置文件;从而得到染色体水平的 scaffolds — groups.asm.fasta。
ALLHiC_plot sample.clean.bam groups.agp chrn.list 500k pdf
Input files: **bam file - mapping bam file **agp file - Placement of contigs in each Hi-C groups, which can be generated by ALLHiC_build **chrn.list - a list of group name and length. The format of this file is like below: ## 第一列为 组名,第二列为对应的长度 group1 125000 group2 159000 **bin size - heatmap bin size ## 热图大小 **ext - extension of plot figures, e.g. pdf ## 输出格式