基因组组装评估 --- Merqury
Background
如今通常使用 Illumina reads 进行与 assembly genome 比对,以评估推断基本水平的准确性,然而对齐短 reads 时,一致性的碱基很容易被检测为变异(SNP 或小插入缺失);此外,这种方法严重依赖于短 reads 比对。单拷贝直系同源物(BUSCO)被广泛应用于评估组装的基因含量,用于测量组装完整性和错误重复。但是 BUSCO 局限于已被广泛研究的物种,而对于新组装的基因组,分析可能不准确,毕竟 BUSCO 是基于目标物种同源基因。此外,BUSCO 只检查保守的单拷贝基因,而未能评估基因组中最难组装的区域。
相较于以上两种评估基因组的方法,k-mers 可以以无参考基因组的方式用于评估基因组组装质量指标。二倍体组装一般生成代表两个单倍型的 primary 和 alternate 组装。primary 通常是一个假单倍型,它捕获纯合区域以及杂合等位基因的单个拷贝,这种伪单倍型不能保证长距离的定相,因此为了估计相位块统计,必须将 alternate 等位基因映射回 primary 组装以确定对应于 primary-alternate 单倍型相位块的区域。
Merqury 仅使用 k-mers 生成组装评估指标。Merqury 将来自未组装的高精度测序 reads 的一组 k-mer 与基因组组装进行比较以进行评估。Merqury 建立在 KAT 引入的基于 k-mer 的分析之上,但增加了用于评估二倍体基因组分型组装准确的新功能。Merqury 生成的指标有 一致性质量(QV) 和 k-mer 完整性。当亲本基因组序列可用时(组装或未组装),Merqury 可以输出单倍型完整性、相位块统计、切换错误率以及可视化表示子代基因组的相位一致性。
这里再介绍一种特殊的 k-mer — hap-mer,hap-mer 定义为单倍型特异性 k-mer,它仅在基因组中单个单倍型上出现一次或多次。Merqury 将 hap-mer 识别为一组遗传的、父母特异性的 k-mers。hap-mers 用于确定 Merqury 中的相位块,其中块被定义为源自相同单倍型的一组一致的标记。可视化每个单倍型组装中的 hap-mers 可以更加直观的表现整体相位一致性。
使用 k-mer 计数光谱, Merqury 可以揭示组装中的拷贝数错误,并准确测量组装完整性和一致性质量。当亲本 k-mers 可用时,Merqury 还可以测量定相精度和单倍型完整性。
k-mers:长度为 k 的基因组子串。
BUSCO:https://academic.oup.com/bioinformatics/article/31/19/3210/211866?login=true
KAT:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5408915/
hap-mer:单倍型特异性 k-mer。
Dependency
- gcc v7.4 或者更新版本
- meryl v1.3
- java(JRE)
- R v4.0.3+ with argparse、ggplot2、scales
- bedtools
- samtools
Github Path:https://github.com/marbl/merqury
Installation
不推荐这种直接安装的方法,因为很难兼容各种程序的依赖,所以我们选用简单的 conda 进行配置运行 Merqury 所需的环境。
为 Merqury 创建一个独立的环境,运行 conda create。
1 | conda create -n merqury -c conda-forge -c bioconda merqury openjdk=11 |
当需要启用 merqury 环境,只需运行 conda activate merqury。
通过 conda 创建 merqury 环境,全文提到的 $MERQURY 为 $home/miniconda3/envs/merqury/share/merqury/
Run
1 | ln -s $MERQURY/merqury.sh # Link merqury |
< >:必需的
[ ]:可选的
mat.meryl — 来自母本单倍型的 k-mer 统计
pat.meryl — 来自父本单倍型的 k-mer 统计
read-db.meryl — 来自 reads 的 k-mer 统计
asm1.fasta — 基因组组装结果
out — 输出文件前缀
Example
1 | ### Download assemblies ### |
只有一个组装(伪单倍型或混合单倍型)
1 | # I don't have the hap-mers |
二倍体有两个单倍型结果
1 | # I don't have the hap-mers |
无需为每个程序集运行 merqury,提供两个 fasta 文件,Merqury 可以为每个文件生成统计信息并合并。
准备 meryl dbs
上面的 Example 是在 *.meryl 存在的情况下运行的;针对自己的项目肯定是需要重新生成属于自己项目的 meryl,故我们有必要学习以下从 0-1 的流程。
获得正确的 k-size
当我们不知道如何设置 k-mer 时,可以运行 Merqury 自带的 best_k.sh 脚本通过提供的 genome_size 生成最佳的 k 值。
1 | sh $MERQURY/best_k.sh <genome_size> [tolerable_collision_rate=0.001] |
使用 meryl 创建 k-mer dbs
Illumina全基因组测序reads
1 | for each read$i.fastq.gz |
10X Genomics全基因组测序reads
构建与合并同常规的
illumina数据,除了需要修剪R1中的前23个碱基。
1 | zcat $input | awk '{if (NR%2==1) {print $1} else {print substr($1,24)}}' | meryl k=$k count output $output - |
构建 hap-mer dbs
这里默认我们已经获得了
maternal/meryl,paternal.meryl和child.meryl。
1 | sh $MERQURY/trio/hapmers.sh maternal.meryl paternal.meryl child.meryl |
或者提交成一个任务:
1 | sh $MERQURY/_submit_hapmers.sh maternal.meryl paternal.meryl child.meryl |
以上会产生以下文件:
1 | * parental specific dbs: `mat.only.meryl` and `pat.only.meryl` |
当我们没有子代 reads,如何创建 child.meryl ?
使用父本和母本的测序集生成的亲本
dbs来制作child.meryl
1 | meryl union-sum output child.meryl maternal.meryl paternal.meryl |
Result
结合组装得到的两个单倍型(二倍体),对组装工具
Gamete binning定相进行评估。
结合自身关注的点,以及拥有的数据,记录 Merqury 对单倍型分型的评估;这一部分的分析只能在 父本、母本、子一代 的 read sets 均可用的情况下进行,并需要创建对应的 maternal.meryl、paternal.meryl 和 child.meryl;具体可参考 hap-mer dbs
除了使用亲本的 read sets 来创建亲本特异性的 k-mers,我们也可以使用亲本的组装结果来生成 maternal.meryl、paternal.meryl。从而与 F1 的 read set 相交生成 hap-mers。
QV estimation(QV 值估计)
假设仅在组装中发现的 k-mers 是 bp 错误,我们可以使用 k-mer 存活率来估计基本水平的 QV。
1 | hap1 2821360 215950548 31.5995 0.000691915 |
- 以上
QV每列的信息:- 分别是两个单倍型与
both - 仅在
assembly中发现的k-mers - 同时在
assembly和read set中发现的k-mers QV值Error rate错误率
- 分别是两个单倍型与
Q30对应于99.9%的准确度;Q40对应着99.99%的准确度;也就是说,得到的两个单倍型计算QV时,其值越高,代表着准确度越高。
Inherited hap-mer plots(继承的 hap-mer plots)
在 F1 read set 上绘制 hap-mer。堆叠版本提供了一个整体视图,未堆叠的线条版本允许直观的确认 hap-mer 分布遵循 二项式分布。预计合理的单倍型的组装将在相应的单倍型组装中包含每个 hap-mers。
https://github.com/marbl/merqury/wiki/3.-Phasing-assessment-with-hap-mers#1-inherited-hap-mer-plots
- spectra-asm plots (显示两个单倍型之间共享的
k-mers)
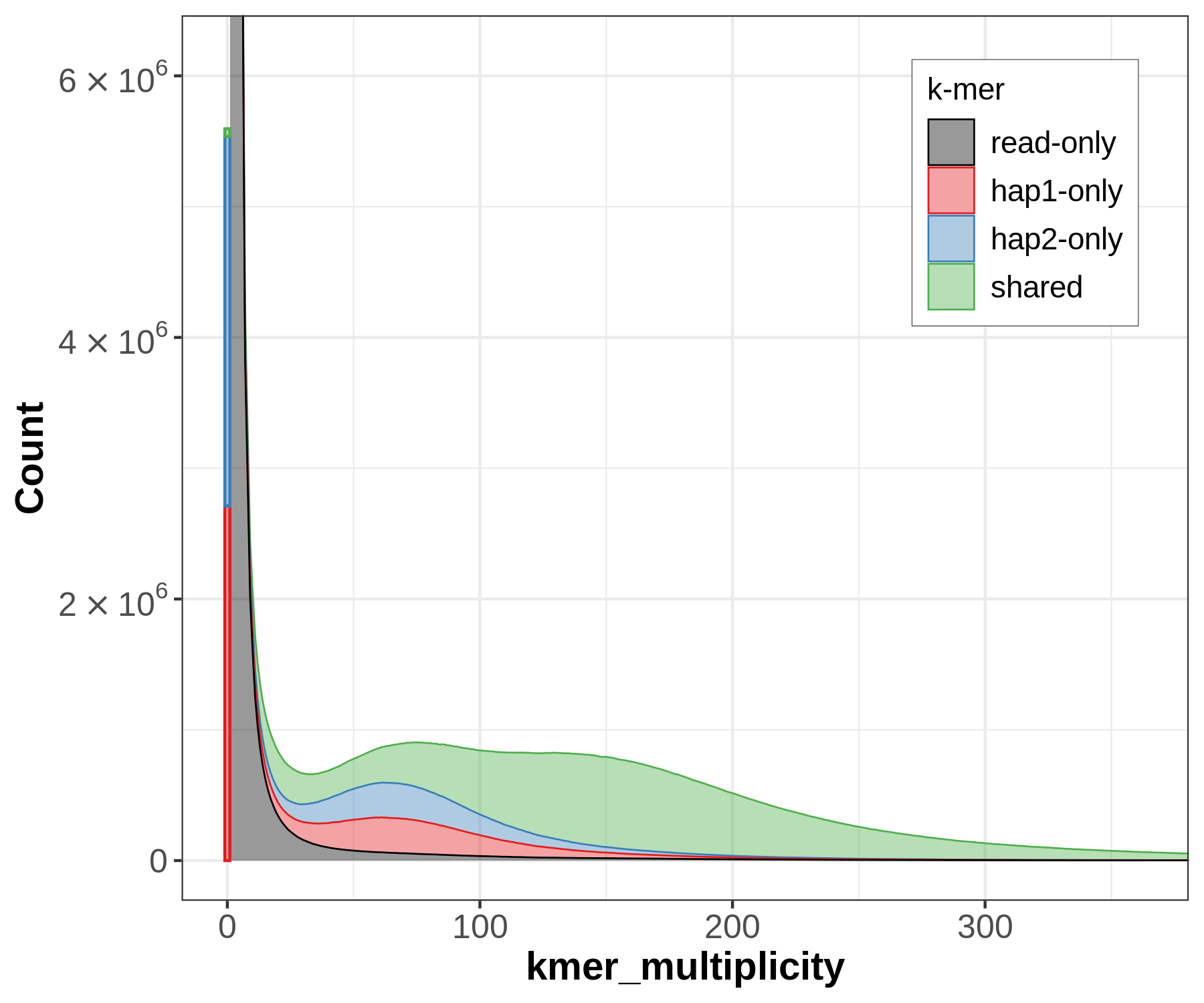
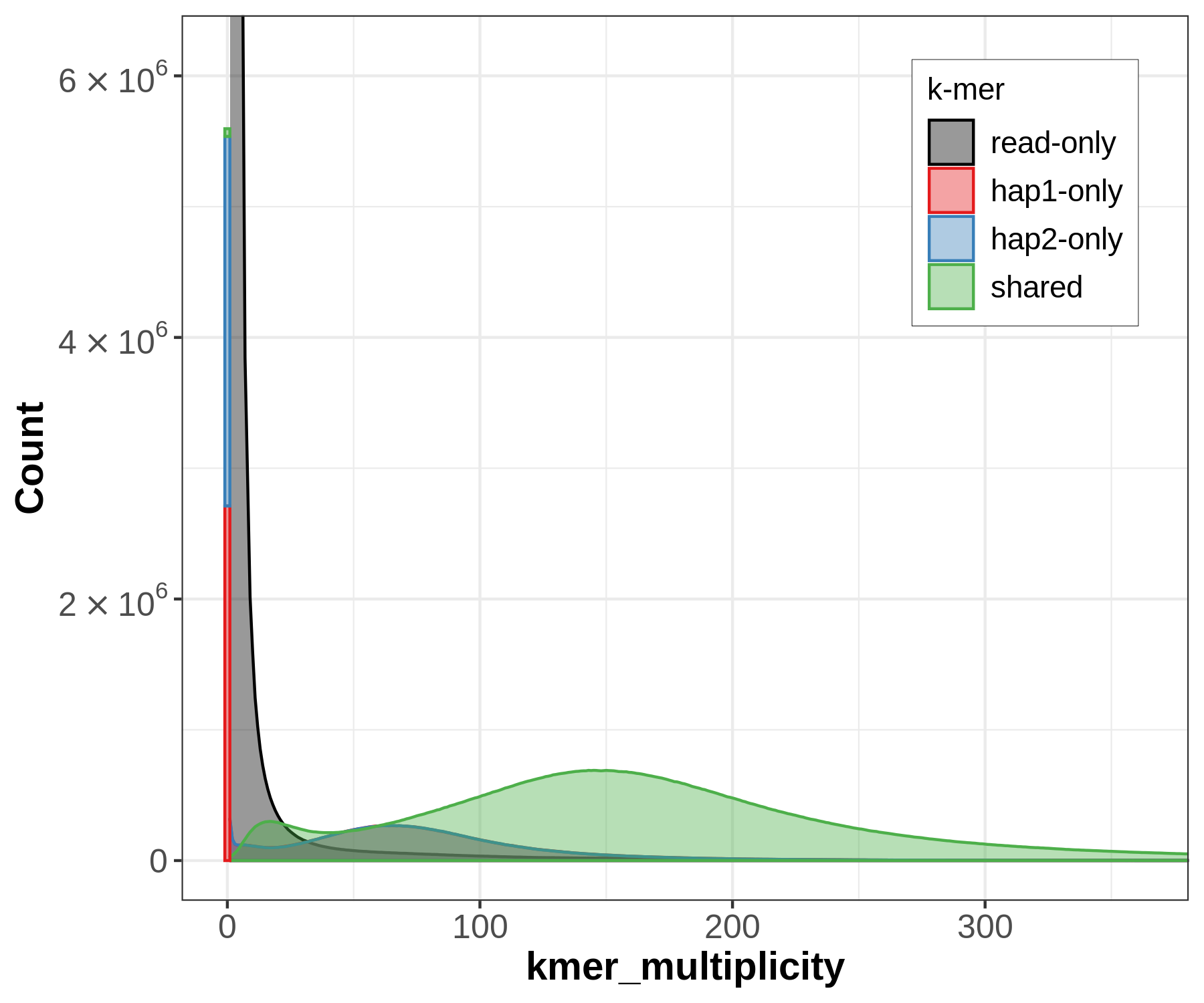
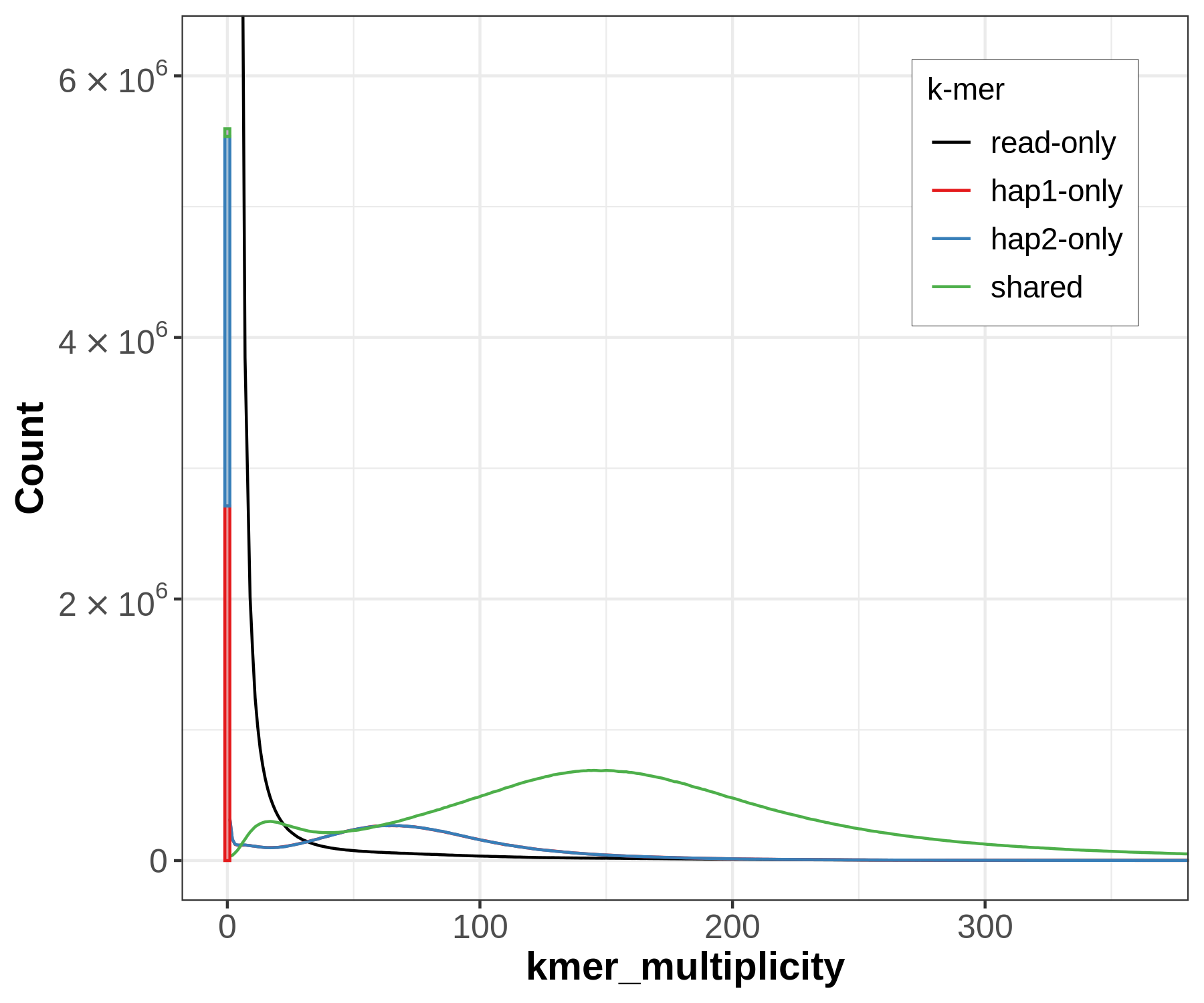
spectra-asm plots用于评估 k-mer 完整性。k-mers 通过它们在 reads 和组装中的存在来进行着色。上图显示,相较于hap1(红色),hap2(蓝色)同样多的k-mers;这里主要看它们的横向是否一致,即覆盖度。若read-only黑色部分出现了峰,则表明组装中缺失的杂合变体。
Hap-mer blob plots(hap-mer 的气泡图)
- hap-mer blob plots (显示两个单倍型的分型效果)
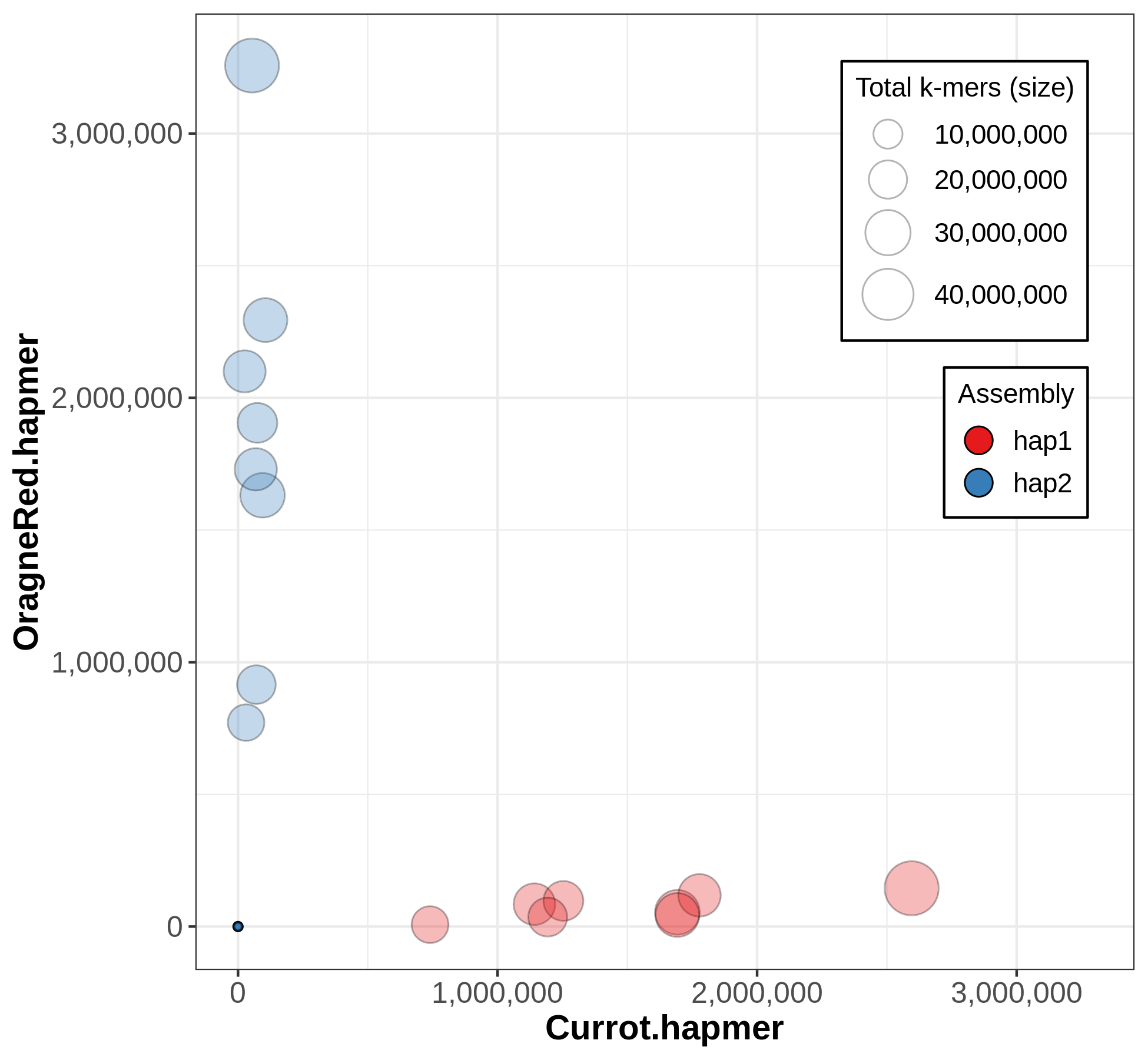
该图显示两个单倍型
hap1和hap2分型效果尚佳,两种颜色的气泡基本都是沿着坐标轴排布的。如出现两种颜色气泡交叉分布,则说明大多数
contigs是来自两种单倍型序列的混合。
气泡图可以直观的体现出整个组装分型的效果如何。图中每一个气泡代表一条序列或 contig 或 scaffold,气泡的大小代表着序列长度。两种不同的颜色对应着不同的单倍型;x 和 y 轴显示对应序列中能找到的 hap-mer 数量。
https://github.com/marbl/merqury/wiki/3.-Phasing-assessment-with-hap-mers#2-hap-mer-blob-plots
相位越多的序列从其他单倍型中发现的 hap-mers 就越少,因此,气泡沿轴排列越近,代表着单倍型定相性能越好。
Hap-mer completeness (recovery rate) 【完整度】
hap-mer 完整性的计算类似于 k-mer 完整性,但这里的是针对每个 hap-mer set。hap-mer 本身已经被过滤;因此,我们可以直接将其用作实际的 k-mers (solid k-mers)。
- 示例:
1 | col col0.hapmer 18316089 18408825 99.4962 |
在这里,我们看到 col 程序集捕获了 99.5% 的 col hap-mers,其中 1% 的 cvi hap-mers 可能是转换错误或碱基对错误。根据从头定相组装策略,这些可能有助于测量组装是否实现了每个单倍型的完全定相。both 则显示了两个组件组合的统计数据,这可以用作衡量整体单倍型完整性的指标。
Spectra copy number analysis per hap-mers(每个 hap-mers 的光谱拷贝数分析)
每个组装与每个
hap-mer生成相关的spectra-cn plots。光谱图可以在一定程度上看出组装结果是否准确,当然这也不是绝对的;但干净的
spectra-cn plots是组装质量的必要不充分条件;也就是说正常的spectra-cn plots也有可能说明组装是错误的,但是不正常的spectra-cn plots说明你的组装不可能是正确的。
- spectra-cn plots (光谱图可以更加直观的显示
k-mers完整性)
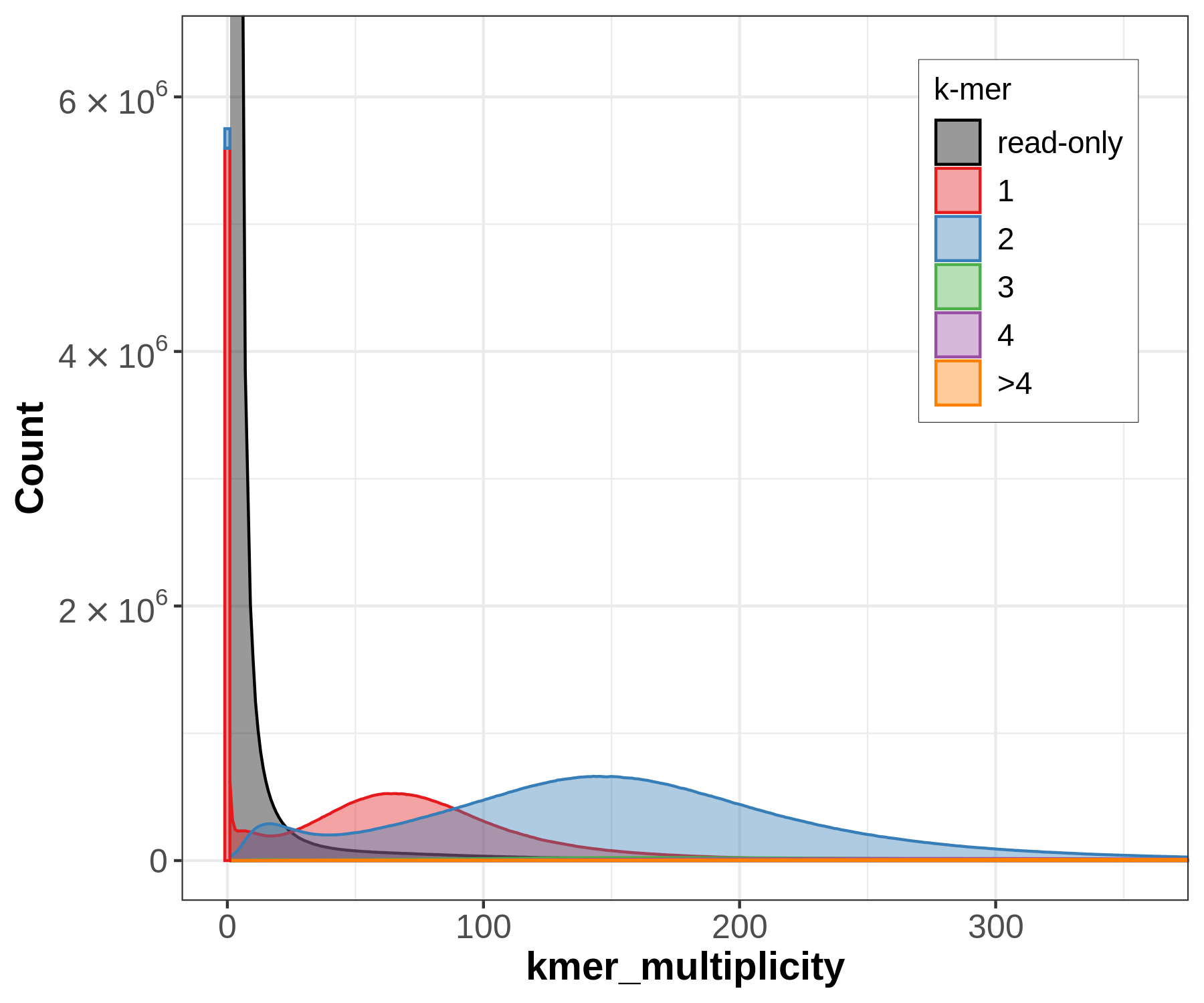
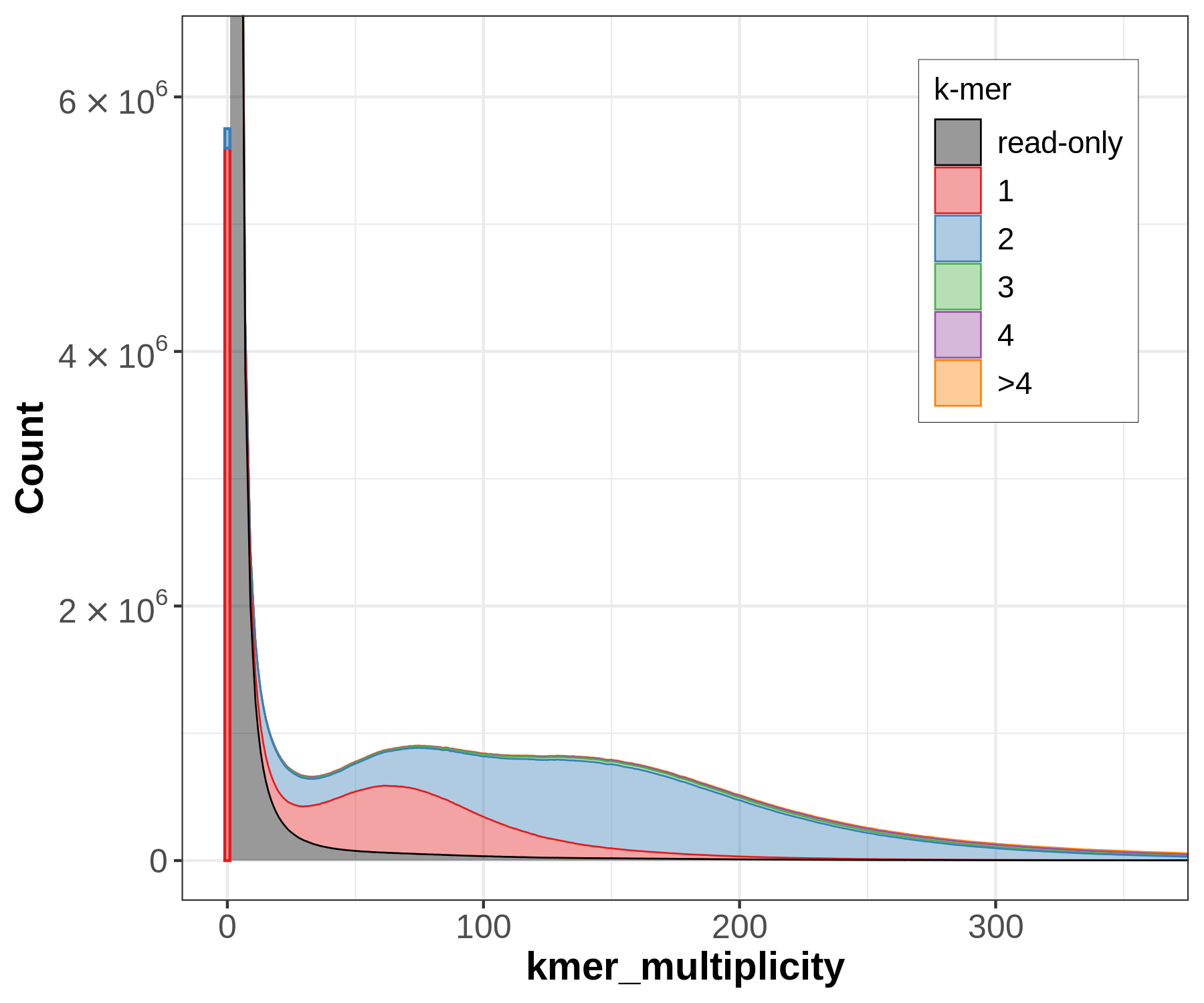
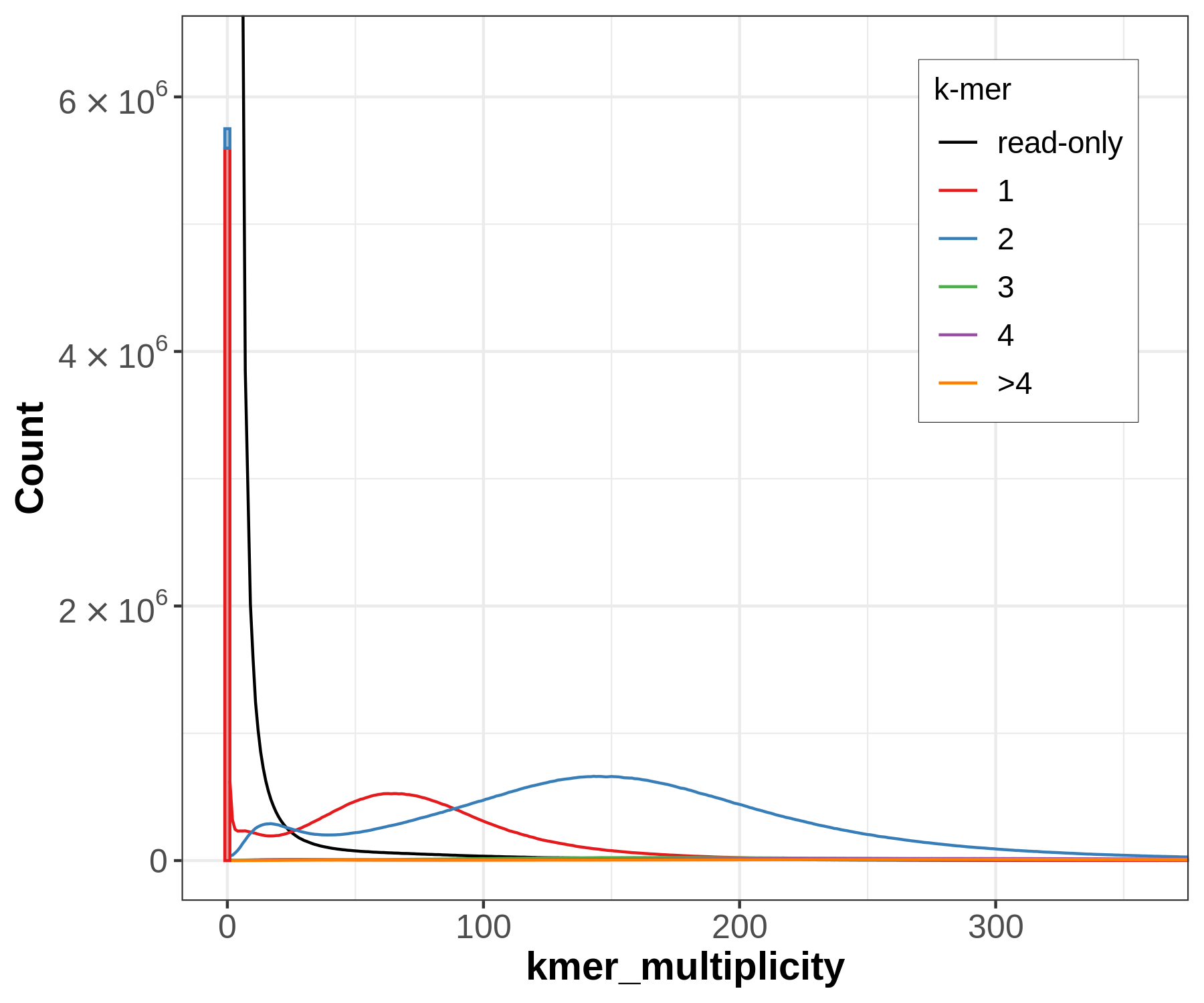
- 以上图分别是:
fl:filledst:stackedln:line
spectra-cn plots旨在 illumina 数据中发现每个 k-mer 的多重性,上图为堆叠/未堆叠直方图,用于可视化组装中每个拷贝数的k-mer计数分布。这里着重关注read-only即黑色部分中低频发现的k-mers,这几乎总是指示着read sets中的测序错误;而在read-only中发现的较高频率的k-mers表明组装中的缺失序列。
Phased block statistics and switch error rates(统计 phased block 和 switch error rates)
phased blocks 由在组装中找到的 hap-mer 定义,通过使用 hap-mers 作为标记,我们可以定义一个 phased block,我们可以在其中看到来自同一单倍型的 2 个以上的标记。
Short-range switches被定义为来自其他单倍型的连续hap-mers,具有确定的序列长度。默认情况下,merqury 将其设置为num_switch: 100和short_range: 20000。
num_switch定义的越大,代表可以容忍更高的short-range switch error rate,从而获得更长的phased block stats。使用更小的num_switch,以获得更多可靠的phased blocks我们得到的越严格,块变得越小,并且,局部的开关延伸形成自己的块。
最终的统计结果在
*.phased_block.stats。
*.phased_block.stats 每列信息为:
- 带有
short-range switch的组装名称。 blocks的数量。blocks中的碱基总数量。- 最小
block的大小。 block的平均大小。blockN50- 最长
block大小。 - 来自其他单倍体的
markers。 blocks中总的markers。Switch error rate。
- 带有
Blocks 在 N* plots 中可视化,按块覆盖度总和的大小排列。块按照单倍型着色,若发现有小块切换到另一个单倍型时,这表明一小部分的 reads 被错误定相。(这可能取决于 long reads 的错误率,预计随着长读错误率的下降而减小 switch error rate。
- phase blocks‘s N plots*(分别显示单倍型中是否存在来自另一单倍型的错误组装)
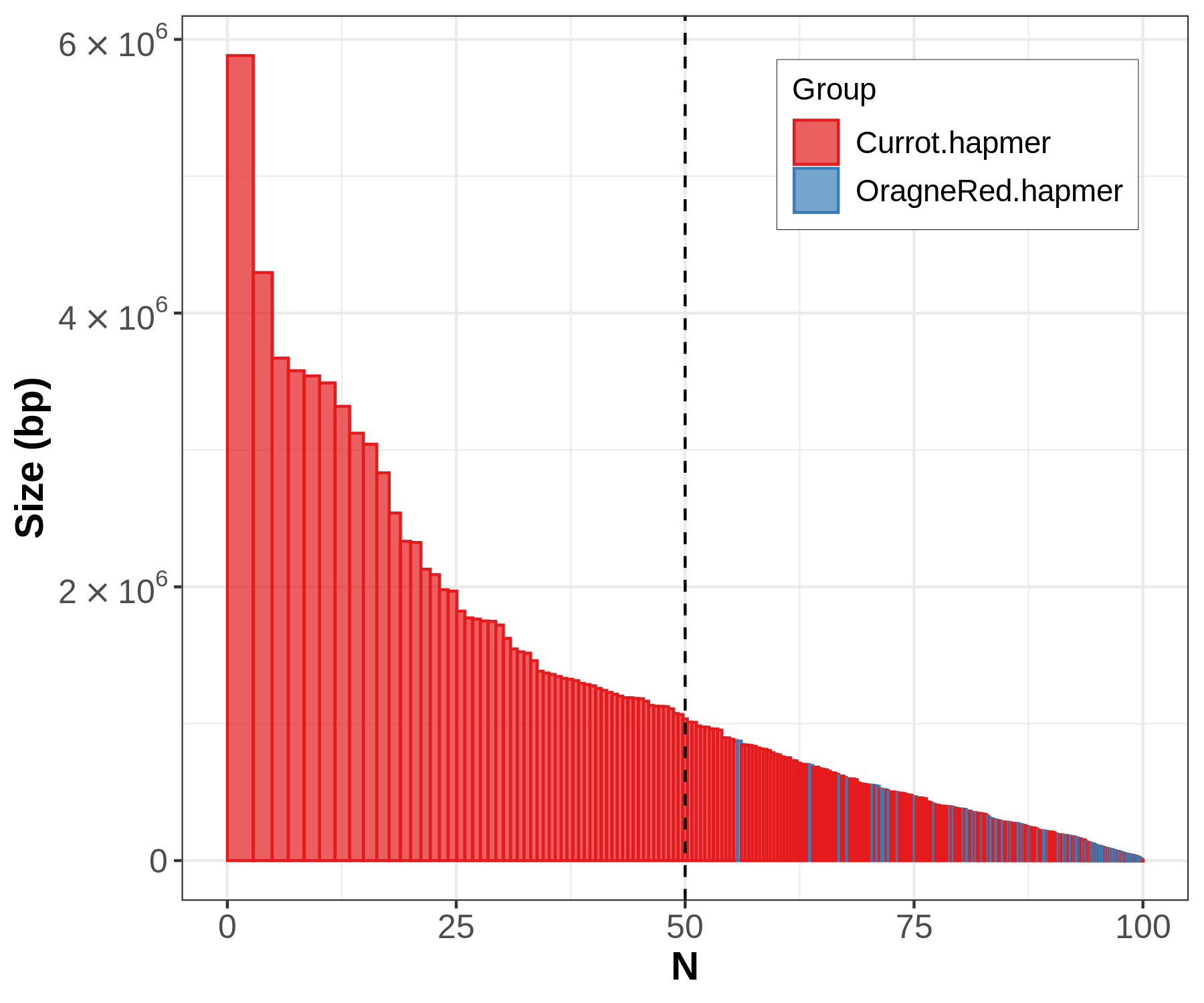
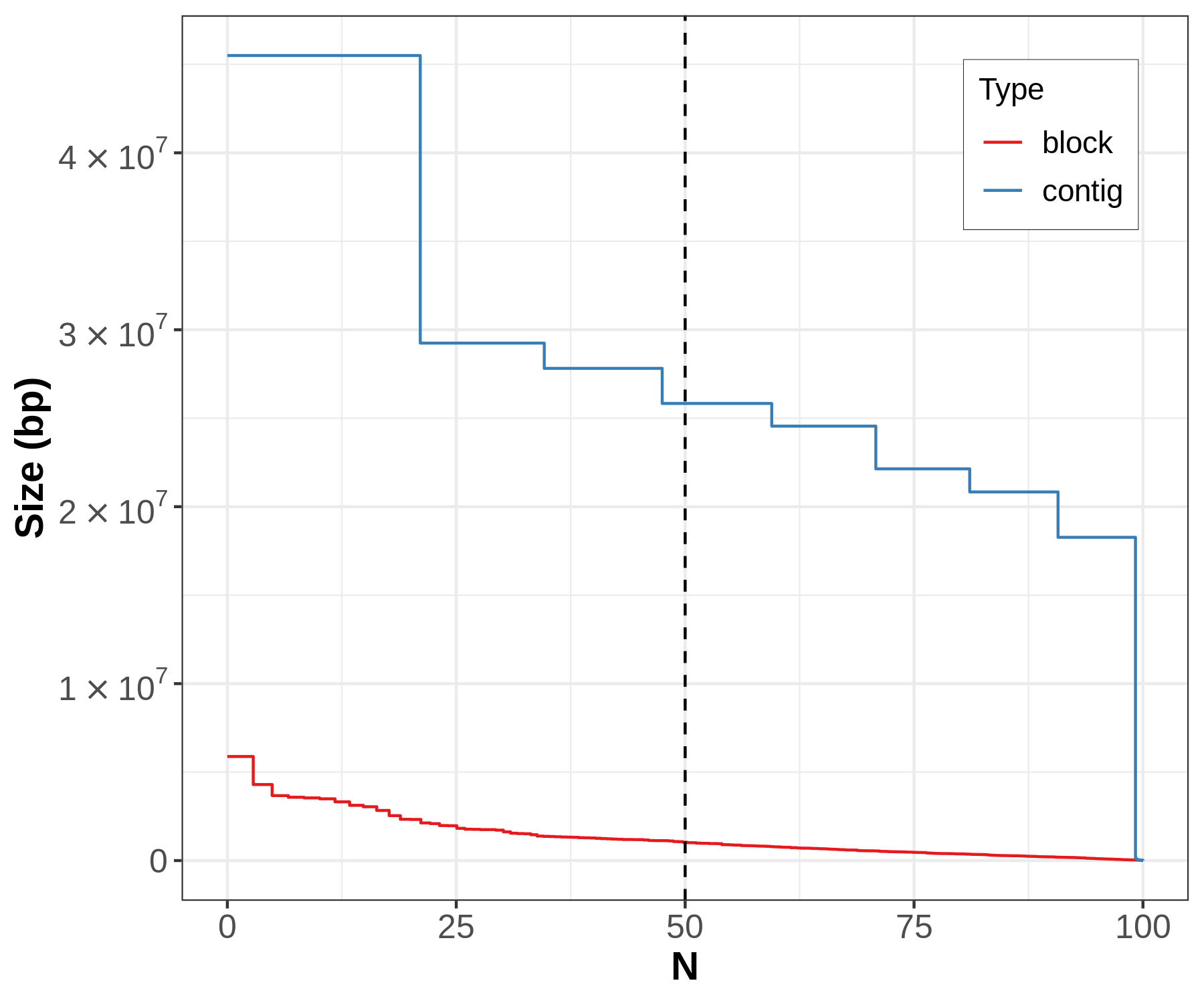
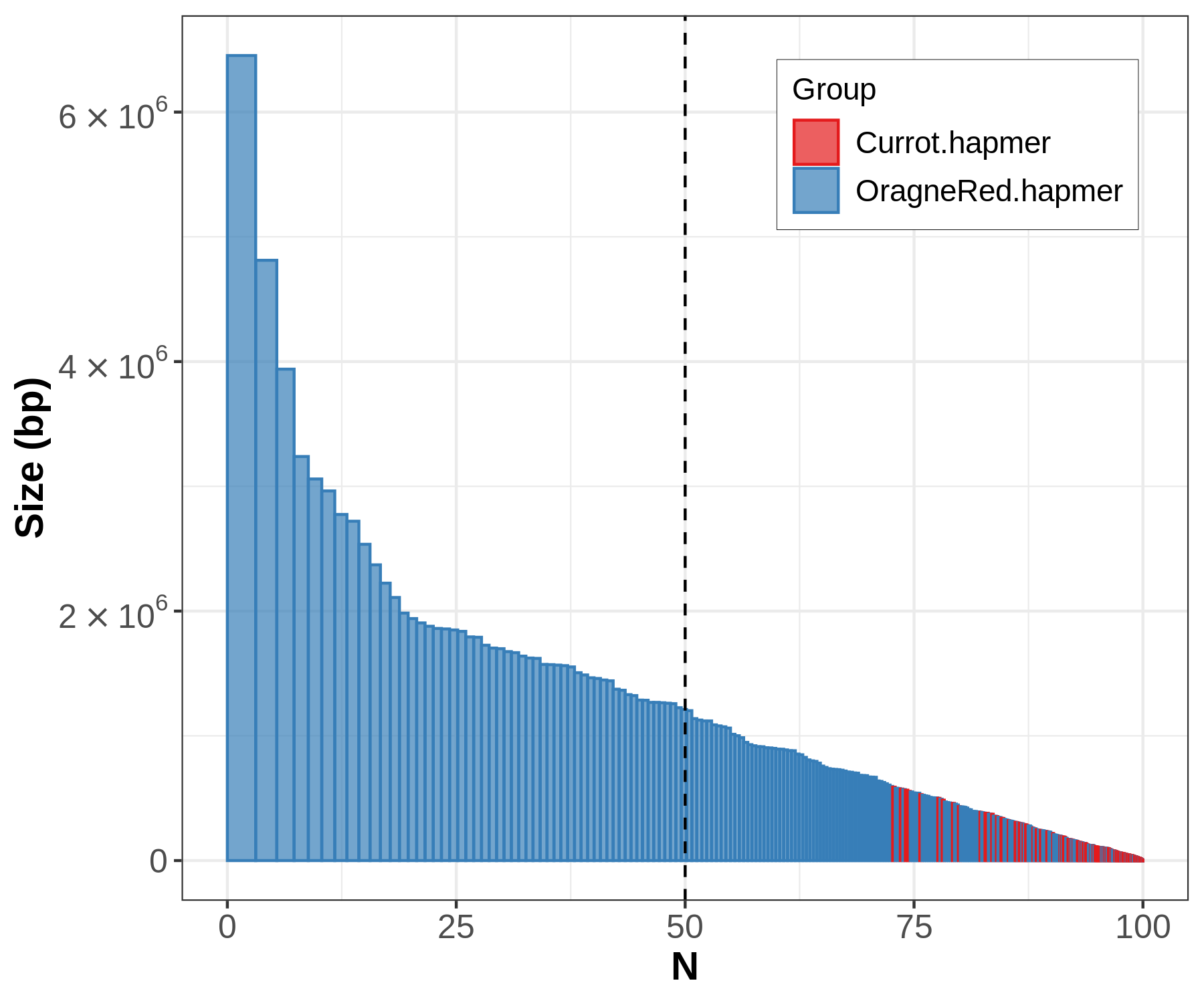
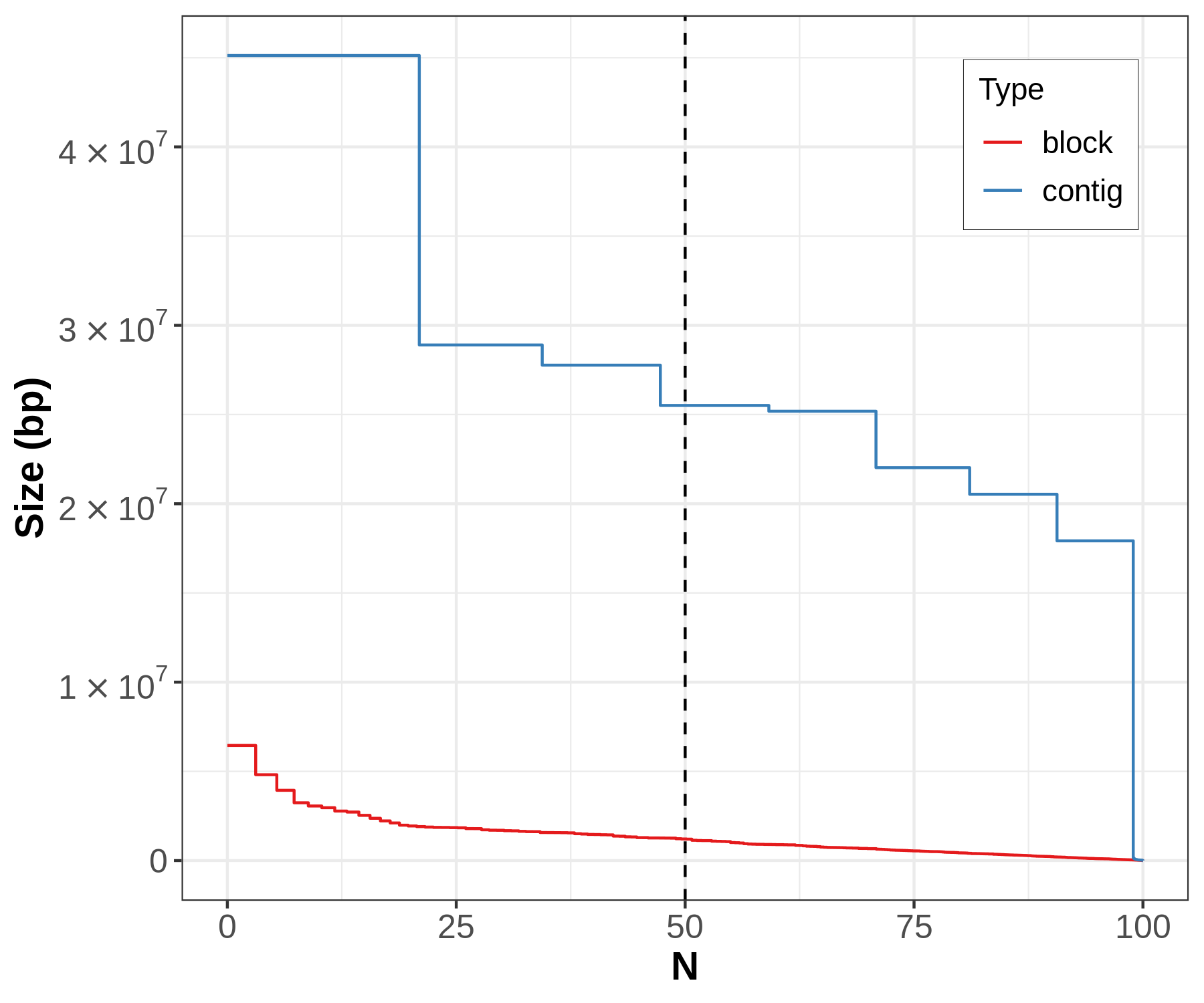
右图可以显示一个组装中两个单倍型组装的相对连续性;上左图显示两个单倍型在
short contigs之间存在彼此的污染;上右两图则显示phase blocks的大小与contigs的大小存在很大的差异,过小的phase blocks表明存在单倍型之间block的频繁转换。
此外,我们还能比较 phased blocks 与 contigs 之间的 N* plots,从而了解重叠群或 scaffold 时如何分型的。
Track each haplotype block in the assembly(跟踪组装中的每个单倍型 block)
Merqury 生成几个可在基因组浏览器 IGV 上加载的文件,以便进一步研究单倍型一致性。
每个单倍型和组装均有:
phased_block.bed
hapmer.bed and hapmer.tdf
References
[1] Rhie, A., Walenz, B.P., Koren, S. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020). https://doi.org/10.1186/s13059-020-02134-9
[2] https://github.com/marbl/merqury/wiki/1.-Prepare-meryl-dbs
[3] https://github.com/marbl/merqury/wiki/2.-Overall-k-mer-evaluation
[4] https://github.com/marbl/merqury/wiki/3.-Phasing-assessment-with-hap-mers